Fatty Acid Oxidation Disorders: Onset, Prognoses, and Impact
By: Jordan Twinkle
Academic, 2017
Abstract
Mitochondrial fatty acid oxidation disorders (FAOD) are a family of rare autosomal recessive diseases [1]. Even though FAOD affect fewer than 1 in 25,000 people [2], they can lead to dangerous or even fatal situations for those afflicted [1]. These diseases are even speculated to be a reason for sudden infant death syndrome (SIDS) giving them serious attention over the last decade. When properly diagnosed, these disorders are truly no threat when managed with diet and supplements. Due to this need for FAOD to be diagnosed, preferably in the perinatal stage, this sector of medicine has received widespread attention in order to combat the growing number of SIDS deaths per year. In future years, research needs to focus on furthering the understanding of the human body’s uptake, trafficking, degradation, and utilization of fatty acids to enable better diagnoses and management of this disorders.
Fewer than 1 in 25,000 people are affected by fatty acid oxidation disorders (FAOD) [2]. I am one of few that has been affected by these disorders, and this influence has driven me to research them extensively. My troubles began as an infant when I experienced infantile hypoketonic hypoglycemia and seizures upon having fat-based immunizations. I struggled with hypoglycemia my entire childhood, without any true explanation for the phenomenon. Upon hitting adolescence, I struggled more and more to keep my blood glucose levels up during intense sports, periods of fasting, and febrile illnesses. By the age of 14, I had severe skeletal muscle egradation and episodic cardiomyopathy. At Riley Hospital’s Molecular Genetics department, I was diagnosed with a FAOD. This completely changed my diet, lifestyle, and career path. Mitochondrial fatty acid oxidation disorders (FAOD) are a family of rare autosomal recessive diseases [1]. Fatty acids are the largest energy reserve the body has for periods of fasting. They provide over 80% of the energy to major organs like the heart, liver, and skeletal muscles especially during times of intense activity. During periods of fast, the body’s glycogen stores become depleted. Fatty acids are broken down through the β-oxidation pathway, releasing energy and ketone bodies which are then used by peripheral tissues and, more importantly, the brain. This energy source is especially important for infants who have a very limited glycogen reserve and a high metabolic rate. This leads to a potentially dangerous and even fatal situation.
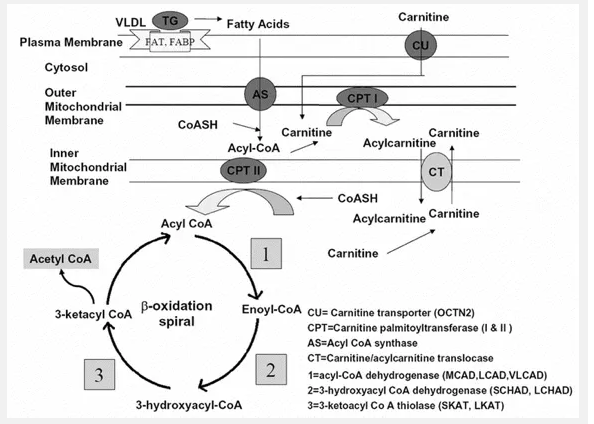
Figure 1. The mitochondrial fatty acid β-oxidation pathway. This diagram shows the substrates and enzymes as well as the location for each step of the pathway. Reproduced from Shekhawat [1].
β-Oxidation Pathway
When the body’s reserve of glycogen starts falling short, the β-oxidation pathway begins, as shown in Figure 1. It all starts with the uptake of fatty acids into the cell. The fatty acids are immediately taken into the mitochondria for degradation. Medium- and short-chain triglycerides are taken into the mitochondria without any aid. The long-chain triglycerides, however, need to be transported by specific mitochondrial membrane transport proteins, such as the fatty acid transporter (FAT). Carnitine plays an essential role in this transportation. Carnitine (3-hydroxy- 4-trimmethylaminobuyric acid) is mainly derived from dietary intake; however, it can also be derived from the degradation of amino acids lysine and methionine [3]. The carnitine is transported into the cell through the OCTN2 transporter [1]. Carnitine palmitoyltransferase I (CPT I) then converts activated fatty acyl-CoA into carnitine esters, and then carnitine-acylcarnitine translocase (CACT) transfers them across the mitochondrial membranes. The fatty acids are then reconstituted by Carnitine palmitoyltransferase II (CPT II). After transportation has been completed, the β-oxidation cycle may officially begin. The first step is the acyl-CoA dehydrogenase reaction catalyzed by flavoprotein-linked enzymes medium-chain acyl-CoA dehydrogenase (MCAD), long-chain hydroxyacyl-CoA dehydrogenase (LCAD), and very-long-chain acyl-CoA dehydrogenase (VLCAD). This step lead to the formation of 2,3-enoyl-acyl-CoA. This is then converted in the second step to produce 3-hydroxyacyl-CoA. This conversion is aided by 2,3-enoyl-CoA hydratase. The third step is converting 3-hydroxyacylCoA to 3-ketacyl-CoA through the two homologous enzymes short-chain hydroxyacyl-CoA dehydrogenase (SCHAD) and long-chain hydroxyacyl-CoA dehydrogenase (LCHAD). The final step involves removing one acetyl-CoA molecule from the 3-ketacyl-CoA by the two homologous enzymes short-chain 3-keto-acyl-CoA thiolase (SKAT) and long-chain 3-keto-acyl-CoA thiolase (LKAT). For long-chain fatty acids, the last three steps of the pathway are catalyzed by the transmembrane MTP. MTP consists of an α and β subunit. The α-subunit contains the LCHAD and hydratase activities, and the β-subunit contains the LKAT activities. The result of the mitochondrial fatty acid β-oxidation pathway is the production of an acylCoA that reenters the cycle and one molecule of acetyl-CoA. Acetyl-CoA can be used for one of three processes: steroidogenesis, enter the tricarboxylic acid (TCA) cycle, or ketone-synthesis. In steroidogenesis, the acyl-CoA is used immediately for the production of steroids. The TCA cycle leads to the production of adenine triphosphate (ATP) directly. If used to synthesize ketone bodies, the FAD-linked dehydrogenases (MCAD, LCAD, and VLCAD) generate electrons from the ketone bodies. These electrons are transferred to ubiquinone by the electron transfer flavoproteins (ETF) and ETF dehydrogenases (ETFDH). This respiratory chain eventually leading to production of ATP.
Diagnosis of FAOD
This is a basic overview of the degradation of fatty acids in the body as an energy source. However, as complete as this seems, much is still a mystery about fatty acid uptake, trafficking, degradation, and utilization. There are many patients who display the typical signs of a FAOD; however, the typical testing done for these disorders still come back negative, failing to show any abnormalities. This means that even though over twenty defects in fatty acid transport and metabolism are known and tested for, some FAODs are still yet to be identifiable. These diseases are even speculated to be a reason for sudden infant death syndrome (SIDS) giving them serious attention over the last decade. When properly diagnosed, these disorders are truly no threat when managed with diet and supplements. Due to this need for FAOD to be diagnosed, preferably in the perinatal stage, this sector of medicine has received widespread attention in order to combat the growing number of SIDS deaths per year. For FAOD, early diagnosis is key for better prognoses. Screening is typically done in the perinatal or infantile stages of life using genetic testing [1]. Typically, genetic testing for a specific FAOD is only done for couples who have a genetic predisposition for a FAOD. Other, less specific tests are done as a simple check for any FAOD. These tests are performed by using mass spectrometry to identify increased levels of acylcarnitine [4]. Waiting at least 24 hours after birth increases the reliability of the test drastically; however, false positives and negatives are still possible. Follow-up genetic testing or blood-carnitine levels are typically the next step.
Specific FAOD Diseases
Of the more than twenty identifiable FAODs, the most common diseases include carnitine uptake defect (CUD), long-chain hydroxyacyl-CoA dehydrogenase deficiency (LCHAD), medium-chain acyl-CoA dehydrogenase deficiency (MCAD), and carnitine transporter deficiency (CTD). [2]. These more common versions will now be discussed for their differences in onset and effects to demonstrate the similarities and vastness of this family of disorders. CUD involves the inability for the affected to properly uptake carnitine from diet alone [5]. Since the diet is the main way for carnitine to be assimilated into one’s cells. The lack of carnitine leads to a buildup of long-chain fatty acids that cannot be brought into the mitochondria and broken-down. The resulting fatty acids build up around the liver, heart, and kidneys. LCHAD is the inability to break-down long-chain fatty acids due to a defect in the dehydrogenase enzyme [3]. This again leads to the build-up of long-chain fatty acids. MCAD is the mutation in the mediumchain dehydrogenase enzyme, causing again a build-up of fatty acids; however, in this case, the medium-chain fatty acids build-up [4]. CTD is the defect in the transmembrane transporter enzyme that first carries carnitine into the cell, making carnitine unable to be used [6]. Again, this leads to the build-up of fatty acids. Though each disorder has the same outcome in terms of the build-up of fatty acids, they all vary drastically in the method they present themselves and the inherited abnormalities that go with it.
Clinical Manifestations
All of the FAOD display themselves through the same symptoms and signs. Though depending on the individual severity of the illness, the disorders can present themselves at different periods of the affected individual’s life [3]. The typical signs for FAOD are episodes of hypoketonic hypoglycemia, changes in behavior, drowsiness, fever, irritability, little appetite, loss of feeling in legs or arms, muscle pain/weakness, vision problems, cardiomyopathy, seizures, and fainting [2]. These present themselves in different fashions, depending on the age of the patient. For infantile ages, metabolic presentation is the most common; FAOD at this age are characterized by bouts of hypoglycemia, poor-feeding, irritability, lethargy, et cetera [3]. These manifestations do not have to be consistent symptoms. They may only present themselves during fasting or illness. For childhood presentation, myopathic results are typically shown. Children usually show the metabolic presentations along with dilated cardiomyopathy, hypotonia, muscle weakness, and elevated creatine kinase (CK) values. The cardiomyopathy in individuals can progress so far that death may occur before diagnosis. If the FAOD has not been diagnosed by the adult stage, typically it is a mild case. The FAOD eventually can come out during extreme illnesses or pregnancies, which both put a lot of metabolic stress on the body. Typically, an FAOD comes out during periods of fasting or illness.
Management
As soon as a diagnosis has been confirmed, patients are encouraged to get genetic counseling and professional dietitian guidance. Most of the complications and symptoms from FAOD can be prevented by maintaining a normal plasma carnitine level. This can be managed by taking oral levo-carnitine (L-carnitine) supplements. These supplements will take care of most symptoms, including hypoglycemic episodes. Hypoglycemic episodes can also be avoided by frequently eating. No special diet needs to be upheld. Dietitians recommend a typical, healthy diet consisting mainly of natural foods, the only difference being the need for small meals to be eaten every 2-3 hours. During times where fasting is needed, like surgical procedures, intravenous solutions of sugar are recommended to keep blood glucose levels within acceptable ranges. Pregnancy for individuals with FAOD can be very taxing; however, with normal supplements of carnitine and a well-maintained diet, no extra precautions need to be taken. In addition to these precautions, cardiomyopathy should be carefully watched for by having annual echocardiograms and electrocardiograms. These can occur every five years upon hitting adolescence. Plasma carnitine levels, once maintained with supplements to a proper level, should be checked annually for all patients to ensure these stay in check. CK levels should be checked during illness to check muscle degradation.
Prognosis
As with any illness, prognosis depends on the severity of the case, the time of diagnosis, and the age of the individual. Treatment with L-carnitine should begin immediately. If it hasn’t irreversible organ damage can occur. The long-term prognosis is very promising as long as carnitine supplementation continues. Individuals who stop carnitine supplementation suffer from cardiac arrhythmia and sudden death from heart problems.
Personal Management
Since being diagnosed with this disorder, I have had to drastically change my lifestyle. I, as suggested, take a L-carnitine supplement three times a day. In addition to this, I take additional medium-chain fatty acids to provide an energy source during times when my glycogen reserves are low since my body has trouble digesting longchain fatty acids. In addition, my body needs to have small meals every couple hours to ensure it stays fueled. Some struggles have occurred due to a lack of consistent energy in my body. When the human body doesn’t have enough energy to go around, it begins to allocate the energy to only the important body functions. Therefore, as a result, the body keeps the circulatory system, the nervous system, et cetera going. However, the immune system, waste systems, et cetera begin to fail. For example, my immune system is often very weak, leaving me ill often and for a long time. In addition, as an athlete in high school, I had a hard time managing my FAOD. During swim try-outs, I passed out after swimming a lap in the pool. During a track meet, I passed out when running a 400m race. During soccer games, I had to be taken out of the game every 10-15 minutes of the 90 minute games to eat or drink something with carbohydrates. Growing up trying to balance a new disease I didn’t understand and high school sports was not easy. I did not understand the importance of my carnitine supplements, so I stopped taking them. My frustration with my disease and my supporting doctors and dietitians pushed me to learn more on my own. Because of my drive to learn more, I began fostering a passion for this field of medicine, the biochemistry of it, and the children, just like me, who want to desperately just run and play like everyone else. I plan to use my technical ability to study medicine at the IU school of medicine. I plan to study endocrinology, molecular genetics, or a related field. My dream would be to work at Riley Hospital for Children at Indiana University Health in Indianapolis to help the children in the area I grew up. I love Indianapolis and the people I grew up loving. Due to my experience with this issue, nothing would give me greater joy in life to help others.
Future Development
Awareness of the importance of carnitine needs to be stressed to individuals with FAOD. Even one individual dying of discontinued carnitine supplements calls the need for better counseling and education. In future years, research needs to focus on furthering the understanding of the human body’s uptake, trafficking, degradation, and utilization of fatty acids to enable better diagnoses and management of this disorders because for FAOD, early diagnosis is the key for a better prognosis. Also, better diagnoses can prevent more SIDS deaths. Due to these reasons, more attention needs to be brought to FAOD, both for the sake of research and public awareness in handling, managing, and properly diagnosing these diseases.
REFERENCES
- Shekhawat, Prem S., Dietrich Matern, and Arnold W. Strauss. “Fetal Fatty Acid Oxidation Disorders, Their Effect on Maternal Health and Neonatal Outcome: Impact of Expanded Newborn Screening on Their Diagnosis and Management.” Pediatric Research. International Pediatric Research Foundation, Inc., 2005. Web. 10 Oct. 2016.
- “Fatty Acid Oxidation Disorders.” March of Dimes. March of Dimes, Jan. 2015. Web. 10 Oct. 2016.
- Magoulas, Pilar L., and Ayman W. El-Hattab. “Systemic Primary Carnitine Deficiency: An Overview of Clinical Manifestations, Diagnosis, and Management.” Bio-med Central. Bio-med Central Ltd., 2012. Web. 10 Oct. 2016.
- “MCAD and Other Fatty Acid Oxidation Disorders.” Genetics and Newborn Screening. Illinois Department of Public Health, n.d. Web. 10 Oct. 2016.
- “Carnitine Uptake Defect.” Genetics Home Reference. U.S. Department of Health and Human Services, n.d. Web. 10 Oct. 2016.
- “Carnitine Transporter Deficiency.” Genetic Fact Sheet for Parents of Fatty Acid Oxidation Disorders. Screening, Technology, and Research in Genetics, 20 Feb. 2016. Web. 10 Oct. 2016.